內(nèi)容速覽
無Ir基氧析出反應(yīng)(OER)催化劑的探索是電解制氫的關(guān)鍵。武漢理工大學(xué)木士春教授等提出了一種Se,S異陰離子調(diào)控策略����,通過誘導(dǎo)的結(jié)構(gòu)不對稱性來提高陽離子Ru位點的OER活性和穩(wěn)定性�。由于Se和S在原子半徑�、金屬量和電離能上的差異,它不僅改變了原始RuS2的八面體排列和鍵長�����,而且還充當電荷調(diào)制器���,促進電子向Ru的積累。令人印象深刻的是�����,電子結(jié)構(gòu)和配位環(huán)境優(yōu)化的Ru2(S3Se)實現(xiàn)了優(yōu)越水氧化活性(186 mV @ 10 mA cm-2)���,質(zhì)量活性和比活性分別是商業(yè)RuO2的28.2和10.5倍�。此外,Se和S的協(xié)同效應(yīng)有效地抑制了可溶性Ru物種的形成�����,從而提高了OER的穩(wěn)定性��。理論計算和原位分析技術(shù)進一步揭示了Ru2(S3Se)表面的反應(yīng)路徑和結(jié)構(gòu)變化��,使速率決定步驟(RDS)的能壘降低和Se-Ru-S鍵的強耦合增強OER性能���。
研究主體
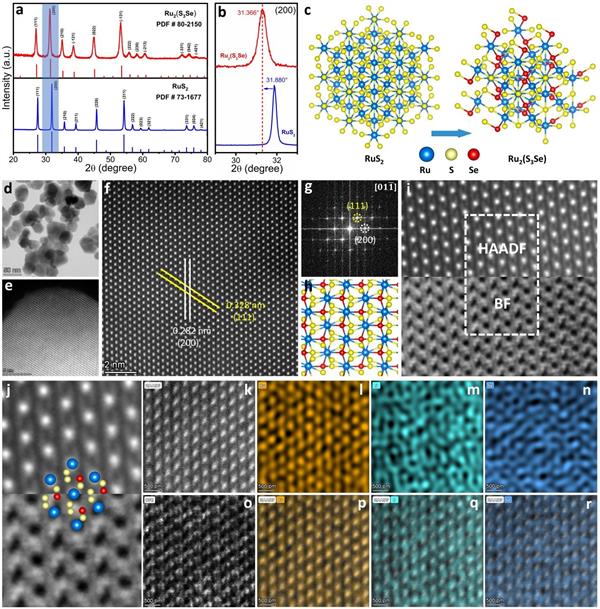
圖1. 催化劑的相識別和微觀結(jié)構(gòu)|(a)合成的Ru2(S3Se)和RuS2的XRD圖�;(b)圖a中藍色陰影部分的放大視圖���;(c)RuS2和Ru2(S3Se)的晶體結(jié)構(gòu)����;(d����,e)Ru2(S3Se)的STEM圖像;(f,g)相應(yīng)的高分辨原子圖像和FFT模式;(h)沿Ru2(S3Se)的[011]方向查看的晶體結(jié)構(gòu)�����,Ru、S和Se原子分別為藍色���、黃色和紅色����;(i)Ru2(S3Se)的HAADF和BF-STEM圖像����;(j)圖像i圖像中白框指示的放大區(qū)域;(k-r)STEM圖像以及Ru��、S和Se的相應(yīng)EDS元素圖���。
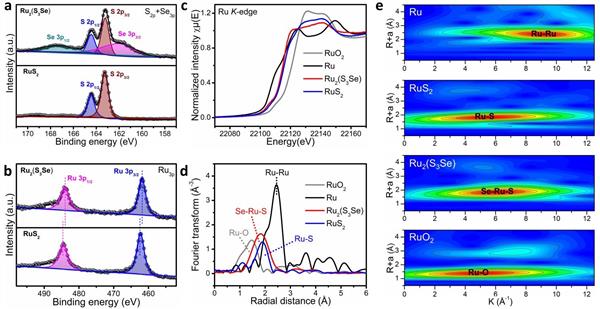
圖2. 催化劑的電子結(jié)構(gòu)|(a) Ru2(S3Se)和RuS2的S 2p和S 3p XPS譜�;(b) Ru2(S3Se)和RuS2的Ru 3p XPS譜��;(c) RuO2����、Ru�、Ru2(S3Se)和RuS2在Ru K邊的XANES光譜;(d) RuO2���、Ru��、Ru2(S3Se)和RuS2的EXAFS光譜的傅里葉變換���;(e) RuO2��、Ru�、Ru2(S3Se)和RuS2的Ru K邊 WT-EXAFS����。
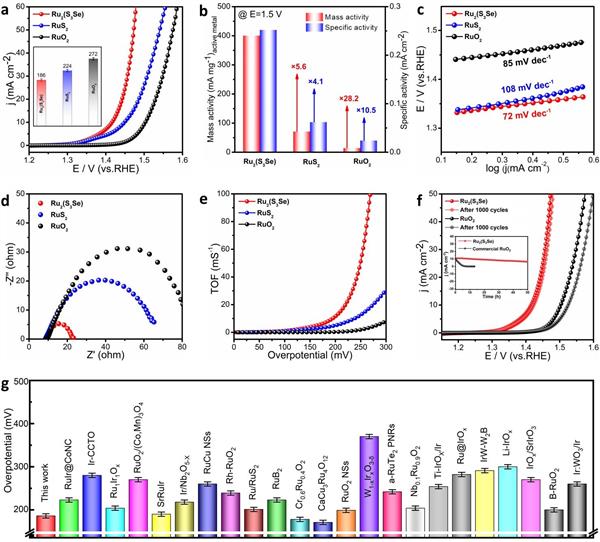
圖3. 酸性O(shè)ER性能|(a)0.5 M H2SO4中Ru2(S3Se)、RuS2和RuO2的OER極化曲線���,內(nèi)插:10 mA cm-2的相應(yīng)過電位�����;(b)Ru2(S3Se)�、RuS2和RuO2的質(zhì)量活性和特定活性����;(c)Ru2(S3Se)、RuS2和RuO2的相應(yīng)Tafel斜率�;Ru2(S3Se)�����、RuS2和RuO2(d)EIS和(e)TOF���。(f)Ru2(S3Se)和RuO2的1000 CV周期前后的極化曲線,插圖:Ru2(S3Se)和RuO2的時間依賴性電流密度曲線���;(g)Ru2(S3Se)的OER性能與最近報道的貴金屬電催化劑的比較���。
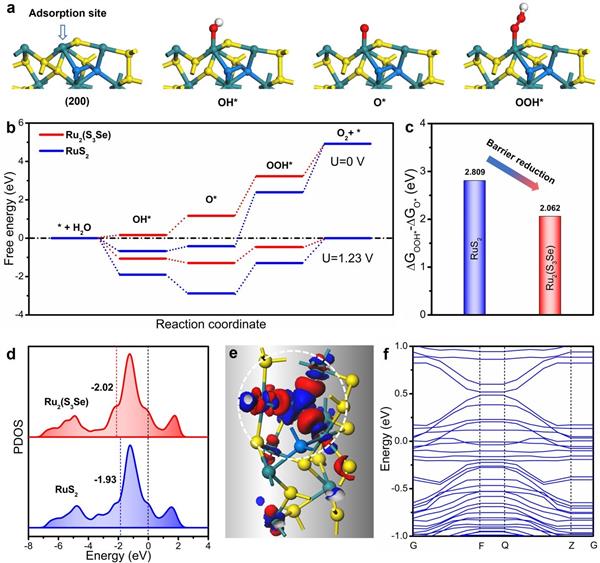
圖4. DFT計算|(a) Ru2(S3Se)吸附位點的四步OER過程;(b)施加過電位分別為0和1.23 V(vs RHE)時�,RuS2和Ru2(S3Se)在OER過程中的自由能分布圖;(c) RuS2和Ru2(S3Se)的PDOS能量變化�����;(d)建成模型上Ru位點的PDOS����;(e) Ru2(S3Se)模型上的差分電荷密度��;(f) Ru2(S3Se)的能帶結(jié)構(gòu)�����。
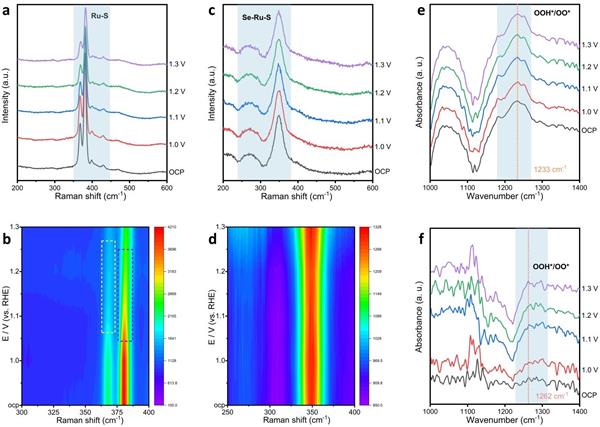
圖5. 原位研究|(a)RuS2和(c)Ru2(S3Se)在OCP~1.3 V vs RHE的電位范圍內(nèi)的原位拉曼光譜;(b)RuS2和(d) Ru2(S3Se)的相應(yīng)輪廓圖��。(e)Ru2(S3Se)和(f)RuS2在OCP~1.3 V vs RHE的電位范圍內(nèi)的原位FTIR光譜�����。
原文索引
Chen, D., Zhao, H., Yu, R. et al. Heteroanion induced structural asymmetricity centered on Ru sites switches the rate-determining step of acid water oxidation. Energy Environ. Sci.(2024).
DOI: 10.1039/D3EE03396A.