三位審稿人一致好評�����!他����,「國家級高層次人才」���,學(xué)科首席教授��,新發(fā)Nature子刊�����!“魚”與“熊掌”亦可兼得��!
四毛&八毛 微算云平臺 2024年10月01日 08:18 廣東
成果介紹
同時活化金屬和晶格氧位點���,通過提供高可用性的活性位點和介導(dǎo)催化活性/穩(wěn)定性來構(gòu)建兼容的多機制催化劑,有望用于析氧反應(yīng)(OER)����,但仍存在重大挑戰(zhàn)。
武漢理工大學(xué)木士春教授等人在OER過程中對NiMoO4·xH2O@Fe,S進行完全重構(gòu)�����,得到Fe和S雙調(diào)制的NiFe氫氧化物(R-NiFeOOH@SO4)�����,并通過原位光譜/質(zhì)譜和化學(xué)探針證實����,在金屬/氧位點同時優(yōu)化的情況下,實現(xiàn)了相容的吸附質(zhì)演化機制和晶格氧氧化機制�。進一步的理論分析表明,F(xiàn)e促進了吸附質(zhì)演化機制下的OER動力學(xué)�,而S激發(fā)了晶格氧氧化機制下的晶格氧活性,表現(xiàn)為O 2p能帶中心上移�、d-d庫侖相互作用放大、金屬-氧鍵減弱���、中間吸附自由能優(yōu)化����。
得益于兼容的多機制�����,R-NiFeOOH@SO4只需要251±5/291±1 mV的過電位就可以在堿性介質(zhì)中獲得100/500 mA cm-2的電流密度���,并且具有超過300小時的穩(wěn)定性能���。這項工作為更好地設(shè)計高性能OER催化劑提供了理解OER機理的見解��。
相關(guān)工作以《Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation》為題在《Nature Communications》上發(fā)表論文�。
值得注意的是����,該工作受到了三位審稿人的一致認可!無論是所提出的新概念��,還是論文所提供的證據(jù)��、結(jié)論都能夠清晰有力地令人信服�����。

眾所周知�,吸附質(zhì)演化機制(AEM)和晶格氧氧化機制(LOM)是常見的OER機制,目前更多的研究主要集中在如何操縱催化劑活性位點以改善�����、促進這兩種機制��,改善催化性能。該研究論文創(chuàng)新地提出了AEM與LOM相容于一個催化劑上����,獲得了驚人的效果!
圖文導(dǎo)讀

圖1 催化劑的制備與結(jié)構(gòu)
通過化學(xué)蝕刻和原位電化學(xué)自重構(gòu)工藝合成了SO42-修飾的NiFe氫氧化物(R-NiFeOOH@SO4)�����,如圖1a所示����。首先�,通過水熱反應(yīng)在泡沫鎳(NF)襯底上生長NiMoO4水合物(NiMoO4.xH2O)納米棒(圖1b),然后將其作為模板在化學(xué)蝕刻過程中引入Fe和S物質(zhì)�。得到的表面粗糙的NiMoO4.xH2O@Fe,S預(yù)催化劑仍主要表現(xiàn)為NiMoO4.xH2O晶體結(jié)構(gòu),結(jié)晶度減弱(圖1c)����。然后,通過陽極循環(huán)伏安法(CV)活化合成R-NiFeOOH@SO4��,去除NiMoO4.xH2O自犧牲模板���。據(jù)此�,通過NiMoO4.xH2O和NiMoO4.xH2O@Fe陽極活化得到R-NiOOH和R-NiFeOOH作為對照樣品��。
FESEM和TEM圖像顯示,所有催化劑都繼承了初始的納米棒結(jié)構(gòu)(圖1d�、e),R-NiFeOOH@SO4在表面表現(xiàn)出明顯的顆粒細化�。活化后���,R-NiFeOOH@SO4的BET表面積增加����。HRTEM圖像(圖1f)揭示了NiMoO4?xH2O納米線的高結(jié)晶度��,而NiMoO4.xH2O@Fe,S(圖1g)和NiMoO4?xH2O@Fe存在明顯的表面非晶化和更多的晶格缺陷���,進一步證實了蝕刻導(dǎo)致的結(jié)晶度下降���,這更有利于OER重構(gòu)。在陽極CV活化后�,HRTEM圖像中幾乎看不到屬于初始NiMoO4.xH2O的晶格間距(圖1h),晶格間距0.208和0.240 nm分別對應(yīng)于NiOOH(210)和(011)晶面���,確定了NiMoO4.xH2O模板成功轉(zhuǎn)化為NiOOH活性物�。此外,EDS(圖1i)證實了所有催化劑中Mo元素幾乎完全去除�����,而Ni�����、Fe����、S和O元素均勻分布在R-NiFeOOH@SO4的整個納米棒區(qū)域����。

圖2 催化劑的電子結(jié)構(gòu)與配位結(jié)構(gòu)
本研究利用XPS和XAS進一步分析了催化劑的電子結(jié)構(gòu)和配位結(jié)構(gòu)。相對于NiMoO4.xH2O, NiMoO4.xH2O@Fe和NiMoO4.xH2O@Fe,S的Ni 2p能譜顯著地向較低的結(jié)合能偏移(圖2a)����,同時Mo的3d結(jié)合能也向較高的結(jié)合能偏移,表明電子從Mo遷往Ni位點��。CV活化后����,所有催化劑的Ni 2p結(jié)合能都出現(xiàn)了異常的負位移,這應(yīng)該是由于在非原位測試下,不穩(wěn)定的NiOOH轉(zhuǎn)化為Ni(OH)2��。但NiMoO4.xH2O@Fe��、S和R-NiFeOOH@SO4之間的負位移較小(-0.24 eV)�����,且R-NiFeOOH@SO4的Ni氧化態(tài)高于R-NiOOH和R-NiFeOOH�,說明Ni2+的轉(zhuǎn)化率較低,這是由于其更有利的完全重構(gòu)過程�。
特別是,在O 1s譜(圖2b)中���,衍生化氫氧化氧的金屬-晶格氧(M-O)信號表現(xiàn)出明顯的正偏移�����,這表明晶格氧(M-O(2-δ)-)的氧化參與了電化學(xué)活化過程��。其中�����,R-NiFeOOH@SO4的M-O偏移更為顯著(0.4 eV)����。同時,與NiMoO4?xH2O@Fe,S相比����,R-NiFeOOH@SO4的氧空位含量由于OH-在電解液中的再填充而降低。此外�����,R-NiFeOOH@SO4的Fe 2p信號峰在圖2c中向更高的結(jié)合能移動���,表明表面Fe物質(zhì)向更高的氧化態(tài)轉(zhuǎn)變,而R-NiFeOOH的Fe 2p結(jié)合能保持穩(wěn)定在+3價����。如圖2d所示,R-NiFeOOH@SO4的S 2p信號完全轉(zhuǎn)化為氧化S物種�,對應(yīng)于硫酸鹽(SO42-)的特征信號峰。
Fe���、S的引入對NiMoO4.xH2O的Ni位點電子結(jié)構(gòu)的調(diào)整可以通過Ni的K邊XANES光譜進一步證實����。CV活化后,所有催化劑的吸收邊都向低能方向移動����,其中R-NiFeOOH@SO4的移動程度較低,與R-NiOOH和R-NiFeOOH相比��,位于能量最高的位置(圖2e)���,與XPS分析結(jié)果一致��。與NiMoO4.xH2O@Fe相比���,F(xiàn)e的K邊XANES(圖2f)在NiMoO4?xH2O@Fe,S中也顯示出較低的平均Fe氧化態(tài),并在R-NiFeOOH@SO4中向高能量轉(zhuǎn)移��。此外�����,S的K邊XANES光譜證明R-NiFeOOH@SO4中的S物種以SO42-的形式存在����,進一步揭示了S物種的氧化。
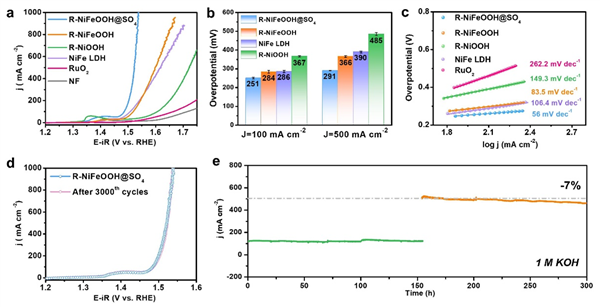
圖3 電催化OER性能
在1 M KOH中�,最優(yōu)R-NiFeOOH@SO4只需要251±5 mV的OER過電位就可以實現(xiàn)100 mA cm-2的電流密度���,低于R-NiOOH(367±4 mV)、NiFe-LDH(286±6 mV)和R-NiFeOOH(284±7 mV)(圖3a�����、b)��。特別是R-NiFeOOH@SO4可以在291±1和308±1 mV的過電位下獲得500和1000 mA cm-2的高電流密度����。如圖3c所示,它還具有最低的Tafel斜率(56 mV dec-1)和最小的電荷轉(zhuǎn)移電阻(Rct)��,表明其具有良好的OER動力學(xué)和高效的電荷轉(zhuǎn)移���。
另一方面,對于R-NiFeOOH@SO4(圖3d)����,在3000次循環(huán)CV掃描前后均未觀察到明顯的衰減,甚至在100和500 mA cm-2下穩(wěn)定運行300 h����,僅略有衰減(圖3e)���,充分說明了OER催化穩(wěn)定性的顯著性。經(jīng)過長期穩(wěn)定性測試����,通過SEM、TEM���、EDX�、拉曼和XPS表征����,R-NiFeOOH@SO4的納米珊瑚狀結(jié)構(gòu)和組成仍然保持良好,證明了其堅固的結(jié)構(gòu)框架���。
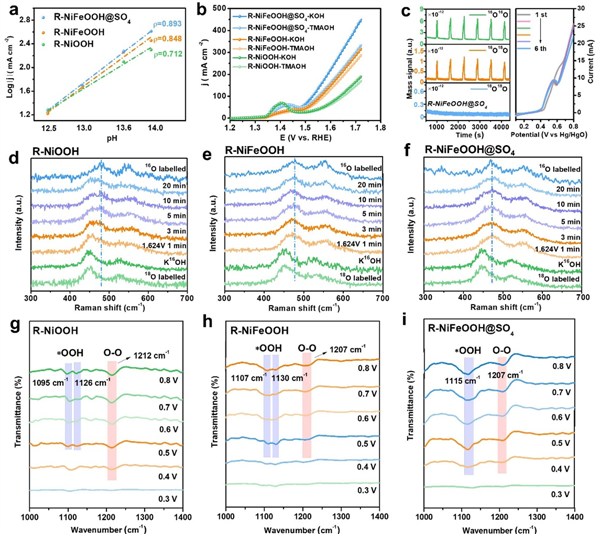
圖4 兼容AEM-LOM的OER催化分析
本研究進一步探討了R-NiFeOOH@SO4參與的協(xié)同催化機制和OER活性的來源��。在圖4a中���,所有催化劑的OER活性都表現(xiàn)出一定程度的pH依賴性,而R-NiFeOOH@SO4的電流密度隨著pH值的降低而下降得更快�,這說明晶格氧參與了OER過程。此外��,R-NiFeOOH@SO4在1 M TMAOH中表現(xiàn)出明顯的OER活性減弱,這是由于在四烷基銨陽離子(TMA+)的強結(jié)合下抑制了LOM���,而它對R-NiFeOOH和R-NiOOH只有輕微的活性衰減(圖4b)���。
為了進一步驗證催化劑晶格氧在OER過程中的參與,采用了原位18O同位素標記微分電化學(xué)質(zhì)譜(DEMS)方法���。催化劑被標記18O�����,以進一步檢測在含有16O的電解質(zhì)中OER過程中O2的釋放����。如圖4c所示���,所有18O標記催化劑的DEMS測量結(jié)果均呈現(xiàn)m/z=32和m/z=34的信號����,表明16O2和16O18O氣體的生成���,證實了有一個晶格氧參與了釋放O2�。未經(jīng)18O標記的R-NiFeOOH@SO4的DEMS結(jié)果只顯示16O16O信號�����,沒有檢測到明顯的16O18O��。同時��,與R-NiOOH相比�,R-NiFeOOH的16O18O峰面積增大,而其16O18O/16O16O的面積比減小�����,說明在AEM途徑下����,F(xiàn)e調(diào)制對析氧的貢獻更為顯著。
此外�,用18O同位素標記的原位拉曼光譜也證實了晶格氧參與了OER過程。如圖4d-f所示���,在氧質(zhì)量對振動模式的影響下�,18O標記的催化劑明顯向低波數(shù)偏移。在含16O的電解液中��,在1.624 V恒定電位下�,隨著18O的消耗,18O標記催化劑的拉曼峰逐漸移回常規(guī)16O標記催化劑的位置��,其中R-NiFeOOH@SO4晶格氧釋放更快�,拉曼峰在1 min內(nèi)移回。相比之下���,R-NiFeOOH和R-NiOOH需要20 min甚至更長時間才能釋放標記的18O�。這一結(jié)果進一步證實了R-NiFeOOH@SO4在OER過程中更快的晶格氧釋放��,揭示了S物種調(diào)制對晶格氧的有效活化�。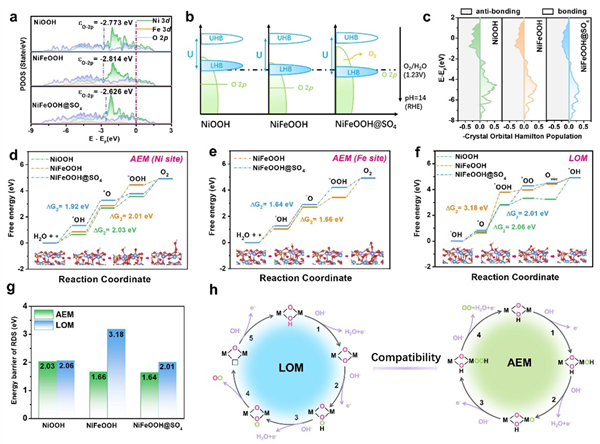
圖5 DFT計算
為了更深入地了解OER協(xié)同機理與活度優(yōu)化之間的聯(lián)系,進一步進行了DFT計算��。選擇實際OER活性物質(zhì)NiOOH����、NiFeOOH和NiFeOOH@SO4作為計算模型。首先�����,計算O 2p軌道的態(tài)密度,以反映圖5a中的晶格氧活度��。與NiOOH(-2.773 eV)和NiFeOOH(-2.814 eV)相比���,NiFeOOH@SO4的O 2p能帶中心(-2.626 eV)相對于費米能級(EF)呈上升趨勢,有利于氧位點電子的移除���,揭示了S調(diào)制對晶格氧的有效活化�����。
如圖5b所示�,計算得到NiOOH��、NiFeOOH和NiFeOOH@SO4的UHB/LHB中心位置分別為1.05/-2.5���、1.12/-2.7和1.14/-2.86 eV�。NiFeOOH@SO4(4 eV)的d-d庫侖相互作用(U)值大于NiFeOOH(3.82 eV)和NiOOH(3.55 eV)���,進一步證實了其有利于晶格氧活化�����。通過計算COHP來評價金屬-氧鍵的強度���。在圖5c中�����,NiOOH�、NiFeOOH和NiFeOOH@SO4的量化Ni-O鍵強度分別為0.752����、0.711和0.666。NiFeOOH@SO4較低的-ICOHP值表明Fe,S共調(diào)制誘導(dǎo)更多的電子被填入反鍵軌道��,導(dǎo)致Ni-O鍵變?nèi)?,助長了晶格氧的參與,并在LOM途徑中形成氧空位�。
在AEM和LOM途徑下,計算了NiOOH��、NiFeOOH和NiFeOOH@SO4的吉布斯吸附自由能圖�。在AEM途徑中分別考慮了Ni原子和Fe原子作為吸附位點的吸附自由能。如圖5d所示���,在Fe,S共改性下�����,NiFeOOH@SO4模型在Ni位點上的RDS能壘優(yōu)化為1.92 eV��。引入Fe后��,*OH在Fe位點的去質(zhì)子能勢壘(RSD)急劇下降���,NiFeOOH和NiFeOOH@SO4的去質(zhì)子能勢壘分別為1.66 eV和1.64 eV,這意味著活性位點轉(zhuǎn)向了Fe(圖5e)���,這從理論上解釋了Fe引入后OER活性的大幅增強����。
LOM的吉布斯自由能圖如圖5f所示�,確定*O-OH的形成為NiOOH、NiFeOOH和NiFeOOH@SO4的RDS��,其能壘分別為2.06��、3.18和2.01 eV�����。計算結(jié)果表明,單一Fe的引入不利于LOM途徑�����,這可能與Fe位點的競爭AEM熱力學(xué)有關(guān)���,而S物種(SO4)的調(diào)制有效地優(yōu)化了LOM途徑中的OER活性����。因此�����,F(xiàn)e�、S共調(diào)制實現(xiàn)了AEM和LOM途徑下對NiFeOOH@SO4的OER中間體吸附的同時優(yōu)化,縮小了AEM和LOM之間的能壘差異�,提高了多機制的相容性(圖5g, h)。
文獻信息
Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation����,Nature Communications,2024.
https://www.nature.com/articles/s41467-024-52682-y